


ABOVE: © istock.com, Andy
Rett syndrome seems to appear out of nowhere. Infants with the rare neurological disorder grow and develop normally at first, but then—generally between 6 and 18 months of age—they suddenly regress. Toddlers forget their words, lose the ability to crawl or walk, develop involuntary hand movements and sometimes seizures, and can even struggle to eat or breathe. The children, predominantly girls, “are very sick,” explains Gail Mandel, a molecular neurobiologist at Oregon Health & Science University who has studied the condition for more than a decade.
The symptoms stem from loss-of-function mutations in a gene near the tip of the X chromosome that codes for methyl CpG binding protein 2 (MeCP2). This protein is a transcription factor, and it’s especially abundant in the central nervous system, where it helps to ensure that particular genes are switched off at the right time during development. “We know a lot about how this protein works,” Mandel explains. “We know how it binds to the genome; we know how it represses genes from being expressed; we know where it is in the nervous system. But what we don’t know is why mutations that eliminate its function give rise to this neurological disease.” That lack of essential knowledge, particularly about how MeCP2 dysfunction affects the expression of other proteins, has hindered attempts at identifying effective drug targets. So, while conducting basic research into that mystery, she says, “I decided to try to think of a way to fix the disease by fixing the mutations in the gene.”
In other genetic diseases, gene therapies have proven successful—and indeed, Mandel gave that idea a go at first. Through multiple preclinical studies, she and her colleagues developed a viral vector designed to slip a healthy copy of the MECP2 gene into cells and get them to make functional MeCP2. But even people with mutations in MECP2 often produce some functional protein, and may even produce normal amounts in some cells. Too much MeCP2 is as bad as too little, so an extra copy of the gene could end up doing more harm than good, says Mandel. “That means that the therapeutic window is very small,” she says.
Researchers involved in RNA editing say it holds numerous advantages over DNA editing.
The gene therapy she helped develop is still being pursued clinically, but what Mandel says she really wants is a way to repair the mutation without increasing the overall abundance of the protein. So she decided to try correcting the messenger RNA (mRNA) for the protein, using an enzyme that can switch out certain mutated bases in transcripts with the correct one. In a paper published in 2020, she and her colleagues successfully used this approach to restore MeCP2 function in live mice with a specific mutation in MECP2.
Mandel sees so much potential in RNA editing that she cofounded VICO Therapeutics, a biotech startup centered around the technology. In 2018, the Rett Syndrome Research Trust awarded nearly $6 million in grants to researchers, including Mandel, pursuing RNA editing–based therapeutics for the condition. Still, Mandel’s 2020 paper was only the second to report successfully employing RNA editing therapeutically in vivo, and there has yet to be a clinical trial to test the approach in humans. In that way, RNA editing-based therapeutics “haven’t even passed the first hurdle yet,” says Mandel, “but the technology is moving super fast.”
In addition to VICO, there are at least seven companies worldwide with proprietary platforms for developing RNA editing–based therapies, although none have yet published preclinical data evaluating those platforms’ efficacy in vivo. Many were already pursuing other nucleic acid therapies, and have recently added RNA editing to their portfolios. Wave Life Sciences, for instance, has developed several oligonucleotides that are being tested in clinical trials, and Beam Therapeutics is known for its DNA base editing platforms; both Massachusetts-based companies are now also pursuing RNA editors.
Increasingly, investment firms and big pharmaceutical companies are taking note of the technology. In August, for instance, Seattle-based Shape Therapeutics signed a $3 billion deal with the pharmaceutical giant Roche, which Shape’s vice president and head of research David Huss says is evidence that RNA editing is taking off. “I think that industry and the larger biopharma companies are really starting to notice that this is an important wave of the future,” he tells The Scientist.
Keay Nakae, a senior healthcare research analyst for the investment bank Chardan, agrees. “From the activity of these announced deals and financings and collaborations, you can see that the interest is heating up.”
Safety first
Editing RNA isn’t all that different from DNA base-editing techniques, which typically use Cas9 or other enzymes attached to a CRISPR guide RNA to replace one nucleotide with another. In the case of RNA editing, the enzymes being used in research are predominantly adenosine deaminases acting on RNA (ADARs), which have multiple functions in humans and many other animals, including ensuring that the cell’s own RNA molecules, which can form double-stranded structures reminiscent of viral genomes, don’t get destroyed by antiviral defenses on the lookout for foreign genetic material. To do this, ADARs flag double-stranded RNAs coming out of the nucleus by converting some of the adenosine (A) bases to inosines (Is), which are read by the cell’s translation machinery as guanosines (Gs).
Researchers have capitalized on this enzyme activity to edit RNA. In the early 2010s, for example, RNA biologist Joshua Rosenthal’s team, then at the University of Puerto Rico, and Thorsten Stafforst’s group at the University of Tübingen in Germany independently combined the editing domain of ADAR with another protein that can attach to a guide RNA, which both provided an attachment point for the protein machinery and transformed the target sequence into a double-stranded RNA ready for ADAR editing (see graphic below). These engineered ADARs and their guide RNAs could be introduced into cells via viral vectors or other means.
Stafforst and Rosenthal published their work in late 2012 and 2013, respectively—just in time to be eclipsed by CRISPR, notes Rosenthal, now at the Marine Biological Laboratory in Massachusetts. “But then, it gradually started dawning on people that, well, if you can change information in DNA, you can change information in RNA as well. . . . So we started getting attention to the site-directed editing of RNA.”
Despite the technologies’ similarities, researchers involved in RNA editing say it holds numerous advantages over DNA editing. Unlike DNA, RNA molecules are transient, lasting only days to weeks in a cell. Even if off-target editing occurs, the edited information doesn’t last forever, so any potential harm is limited in scope, says Rosenthal, a cofounder of RNA editing–based therapeutics company Korro Bio. “RNA editing [falls] somewhere between small molecules, which have a very short duration effect, and CRISPR, which has an almost permanent effect,” he says. “You’ve got to screen carefully these things and look for off-targets—do your due diligence and animal testing and all that. But I think, taken all together in the balance, you’d have to say it’s highly likely that it’s much safer to make off-target edits in RNA than DNA.”
It gradually started dawning on people that, well, if you can change information in DNA, you can change information in RNA as well.
—Joshua Rosenthal,
Marine Biological Laboratory
In addition to its safety advantages, RNA editing could lend itself to a wider variety of clinical applications than DNA editing, Huss says. With CRISPR, “every DNA change will be incorporated into 100 percent of the RNA transcripts that come off of that DNA.” ADARs, on the other hand, typically only edit a fraction of those mRNAs. “You have certain diseases where you may want 100 percent of a change, but you have other ones where you may only want 50 percent, or 70 percent.” One application could be altering mRNA to prevent translation or adjust the structure of tau protein, which helps stabilize microtubules in neurons but is also often associated with neurodegenerative diseases, Huss says. Altering every tau mRNA could cause pathologies, but altering some just might be beneficial.
Partial RNA editing could also prove sufficient for some diseases such as Hurler syndrome, a condition associated with severe skeletal abnormalities, cognitive impairments, and other health problems stemming from a lack of the activity of α-L-iduronidase, an enzyme that helps break down large sugars. About 40 percent of cases are caused by a G-to-A point mutation that creates an improper stop codon. In a recent mouse study, correcting just 30 percent to 40 percent of the mutant mRNAs led to a 60-fold increase in enzyme activity, a researcher with the company EdiGene reported in a 2020 conference presentation.
Bringing the tech up to speed
Efforts in the last few years have focused on increasing RNA editing’s specificity and efficacy. For instance, Beam Therapeutics is developing the REPAIR™ (RNA Editing for Programmable A to I Replacement) system, which consists of a human ADAR enzyme attached to an inactivated Cas13 enzyme that binds to a guide RNA also introduced into the cell. Initial in vitro tests found that the combo enzyme made very few off-target edits. In a 2020 paper, researchers reported that the REPAIR™ system corrected a mutation that causes cystic fibrosis—specifically, a premature stop codon in the gene coding for cystic fibrosis transmembrane conductance regulator protein. The editing system was able to restore production of full-length protein in multiple cell lines, though the authors noted that the efficiency was low, so further refinements are needed.
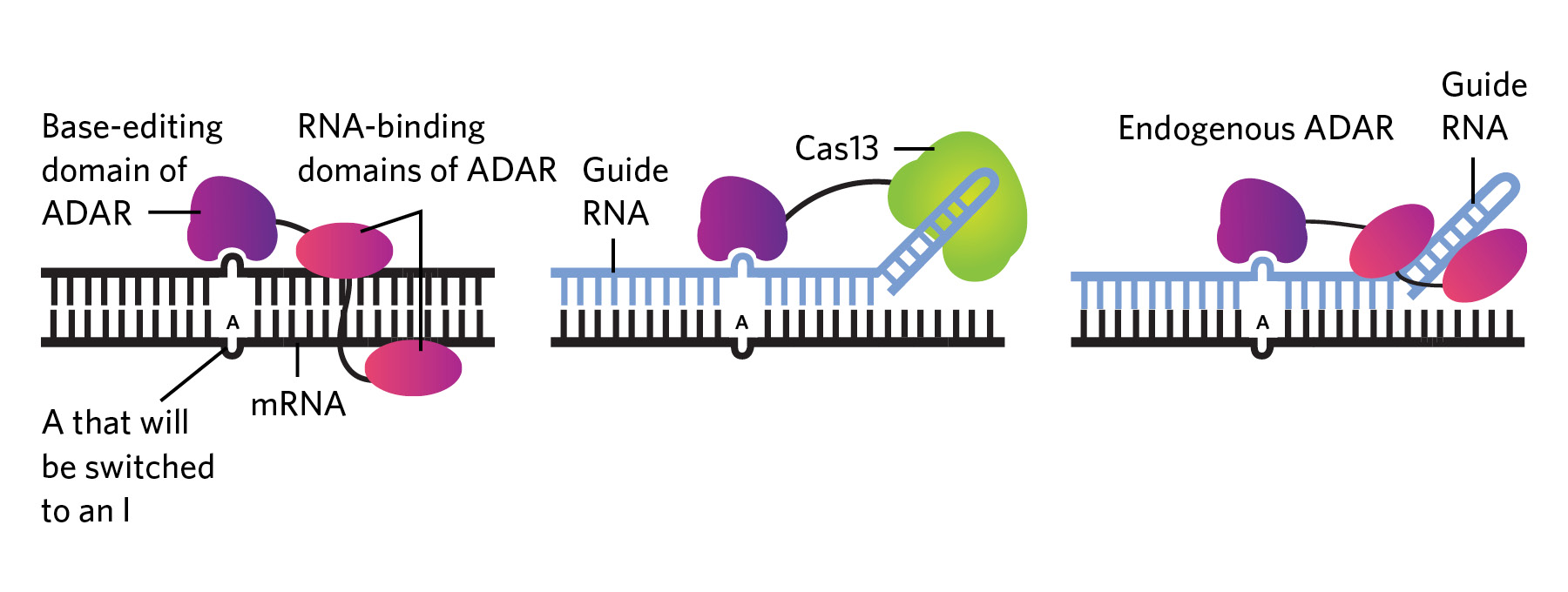
Other companies, such as San Diego–based Locanabio, are also coupling Cas enzymes with ADAR to improve targeting. But there could be disadvantages to using Cas and other bacterially derived proteins, according to Erez Levanon, a computational biologist with Bar-Ilan University in Israel and former consultant for the San Diego company ADARx Pharmaceuticals, which is developing ADAR-based RNA-targeting therapies. Compared to ADARs, which aren’t foreign to human cells, Cas enzymes are more likely to set off immune reactions, he says.
Some groups are tweaking ADARs directly to get the same specificity benefit without using Cas. University of California, San Francisco, biologist Leanna Monteleone and her colleagues, for instance, have developed a unique ADAR that only edits As when there’s no base attached to the ribose backbone across from them in double-stranded RNA—a situation that is extremely rare in naturally occurring RNA, but that can be engineered into a guide RNA. In vitro testing showed that this so-called “bump-hole” strategy led to fewer off-target edits, says Monteleone, who has applied for a patent on the technology.
Now that biotech companies are involved . . . I’m hopeful that things are going to accelerate with RNA editing quickly.
—Gail Mandel,
Oregon Health & Science University
Other teams are exploring the possibility of using a person’s own ADAR enzymes, rather than delivering them alongside a guide RNA. “You can deliver only a guide RNA molecule, and recruit the endogenous wild-type ADAR that’s already present inside the cell, and do that at very high efficiency and specificity,” says Huss—something first demonstrated back in 1995, and something that a few companies, including Shape, are exploring. “That kind of approach is beautiful,” Rosenthal says. “In therapeutics, the simpler the system, the better, in general.” Nakae agrees, adding that the elegance of a system that uses a guide RNA alone to recruit ADARs already present in cells is “one of the reasons why ADAR, I think, is attracting attention.”
Strides are being made on generating guide RNAs, too. Shape Therapeutics, for example, has invested heavily in designing guides, making use of machine-learning techniques and high-throughput screening, says Huss. Such approaches could open up the possibility of editing “any adenosine in the transcriptome”—and with it, the ability to manipulate proteins in novel ways, he adds. Altering A residues involved in splicing reactions, for instance, could promote exon skipping or alternative splicing. RNA editing could also tamp down protein production by converting AUG start codons to GUGs, blocking translation initiation. It may even become trivial to tweak proteins at will—to add or remove a phosphorylation site, for instance, or alter protein cleavage points.
Experts in the field are quick to note that the utility of RNA editing extends far beyond the genetic diseases that have been the focus of biotech interest until now. “The much more nuanced and I think powerful approaches to this down the line are going to be really manipulating systems,” Rosenthal says. His group is looking into the possibility of editing neuronal RNAs to temporarily dull pain, for instance, as an alternative to opiates.
Huss says that one of the big challenges in the near term will be not putting the cart before the horse. “I think the challenge for us is: there’s all these possibilities—how do we focus in on the things that we think will give us the highest probability of success and also prove the technology before we expand it out into many different indications?”
Much to prove
It remains to be seen whether the tech can make good on its potential. Levanon says he expects that, among successes, “there will be many disappointments” in the coming years, especially when it comes to designing efficient guide RNAs. He notes that the bases near the target site on an mRNA, as well as the structure of the guide RNA itself, can tweak the efficiency of ADAR activity, and says that researchers will need to understand more about why natural ADARs edit when and where they do before they can truly manipulate them with precision.
There’s also the obvious limitation that ADARs only edit As into Is. That’s one of twelve possible manipulations one might want to make, notes Levanon; if another base is desired in a given spot, ADARs can’t help. There are enzymes that edit cytosines (Cs) into uracils (Us)—the RNA version of thymine (T)—and these are now being explored, but that technology is years behind, and as of yet, enzymes that make other conversions either aren’t known or aren’t well characterized. While turning an A into an I (read as a G) may be helpful in many cases—in more than half of Rett Syndrome cases, for instance, Mandel estimates—that’s not going to be the case for every genetic disease.
With research at such an early stage, the challenge of drug delivery still looms. “It’s that next step of: Can I deliver the payload to the target tissue? Can I deliver it at a high enough concentration? And can I deliver it so that you have a sustained expression?” that may slow progress toward the clinic, says Huss.
At least in terms of regulatory hurdles on the road to market, “a lot of that ground has already been plowed” by other RNA-based technologies, Nakae says. “I think it’ll actually be an easier path [to approval] than what some of those other modalities faced. But that said, they’ll still have to have the preclinical data that shows that it’s safe before it’s allowed to go forward.” Among companies working on the technology, Nakae says, there are no obvious frontrunners yet.
Nevertheless, “now that biotech companies are involved . . . I’m hopeful that things are going to accelerate with RNA editing quickly,” says Mandel. And she has reason to hope; in May 2021, the Rett Syndrome Research Trust’s press office wrote that RNA editing therapeutics funded by the trust are expected to hit clinical trials by the end of 2024. “It’s certainly an exciting time,” she says.
Correction (December 2): This article has been amended to note that Wave Life Sciences has developed several oligonucleotide-based therapeutics, not RNA-binding proteins. The Scientist regrets the error.


